Болезнь Вильсона-Коновалова (гепатолентикулярная дегенерация) является наследственным нарушением обмена меди в человеческом организме, при котором данный минерал накапливается в различных органах (например, в головном мозге или печени), что приводит к нарушению их функций и патологическим изменениям в них.
Данная болезнь встречается как у женщин, так и у мужчин, примерно 30 случаев на миллион человек. У мужчин заболевание диагностируется в четыре раза чаще, что зафиксировано независимо от места проживания и образа жизни человека. Для регионов, в которых существуют близкородственные браки, болезнь Вильсона-Коновалова диагностируется с большей частотой.
Что происходит в организме человека при нарушении обмена меди? Невозможность поддержания баланса данного элемента приводит к его накапливанию в печени. В это же время происходит блокировка процесса выделения меди с желчью, что соответственно в несколько раз увеличивает её количество. Со временем избыток меди вызывает хроническую интоксикацию, что способствует накоплению элемента в других органах и системах (почках, сердце, головном мозге, костно-суставной системе). В результате происходит токсическое поражение данных органов, что приводит к нарушению их работы.
Причины
Симптом Вильсона-Коновалова представляет собой заболевание, которое передаётся по аутосомно-рецессивному типу: когда один и второй родитель является носителем аномального гена. Причиной возникновения заболевания является мутирование гена, который отвечает за синтез белка, осуществляющего транспорт меди в организме. Данный ген назван АТ-Р7В и находится на плече хромосомы №13. Существует около 80 вариантов мутации этого гена. Самыми опасными мутациями являются те, которые вызывают полное разрушение гена, что приводит к заболеванию в тяжёлой форме.
Манифестирует заболевание в возрасте от 8 до 16 лет, но, несмотря на это почти с самого рождения у больного наблюдается повышенный уровень печёночных аминотрансфераз.
Симптомы
 Болезнь Вильсона-Коновалова может проявляться двояко, особенно различается дебют каждой из форм. Если патология образуется в печени, то со временем развивается цирроз. При неврологических отклонениях поражаются базальные ганглии, кора головного мозга и мозжечок. Невралгическая форма данной болезни характеризуется следующей симптоматикой:
Болезнь Вильсона-Коновалова может проявляться двояко, особенно различается дебют каждой из форм. Если патология образуется в печени, то со временем развивается цирроз. При неврологических отклонениях поражаются базальные ганглии, кора головного мозга и мозжечок. Невралгическая форма данной болезни характеризуется следующей симптоматикой:
- Проявлением тремора и гримасничанья;
- Наличием хореиформных движений конечностями;
- Возникновением атаксии;
- Эпилептическими припадками.
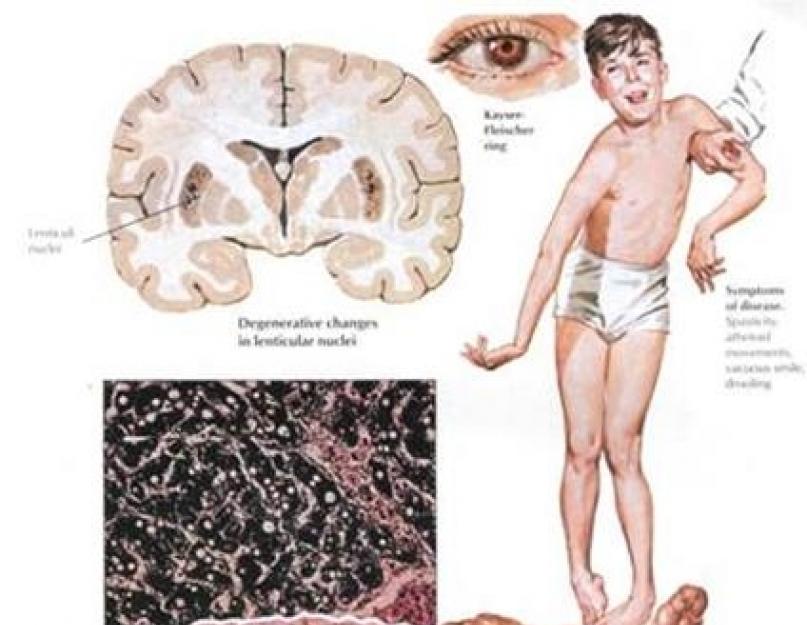
Накопление меди в организме проявляется визуальным эффектом «кошачьих глаз», то есть роговица глаза обрамляется интенсивно окрашенной, яркой окантовкой зеленовато-желтоватого или оранжевого цвета. В медицине данный симптом называется кольцом Кайзера-Флейшера.
Заболевание можно разделить на две стадии:
- Латентная стадия – длиться на протяжении 5-7 лет;
- Стадия клинических (неврологических или печёночных) проявлений.
При остром течении у 25 % больных, болезнь проявляется внезапно, появляется желтуха, повышается температура тела, возникает слабость и расстройство аппетита. Развивается печёночная недостаточность, имеющая молниеносное течение. В большинстве случаев заболевание заканчивается летальным исходом, несмотря на проводимое лечение. Хроническое течение заболевания имеет медленный характер и не проявляется у детей до пяти лет. В 42% случаев болезнь Вильсона-Коновалова начинается с процесса поражения печени.
Печёночные и неврологические нарушения появляются с одинаковой частотностью и при отсутствии своевременного лечения возможно развитие обеих форм болезни. Начальная стадия характеризуется лишь психическими нарушениями, которые даже при длительном лечении не искореняются. Часто дебют заболевания сопровождается следующими симптомами:
- Лихорадка;
- Поражение суставов;
- Гемолитическая анемия.
От переизбытка меди в организме, характерном для синдрома Вильсона-Коновалова, возникает целый ряд сопутствующих заболеваний: сахарный диабет, аневризм, патологическое изменение сосудов, развитие атеросклероза и синдрома Фанкони.
Видеоматериал по теме:
Диагностика
Диагностика начинается с физикального обследования, которое позволяет выявить визуальные патологические симптомы болезни. Далее при помощи лабораторных исследований мочи и крови больного позволяет определить наличие повышенного уровня печёночных ферментов и суточное выделение меди в урине.
Также, для постановки точного диагноза, необходимо использование инструментальных методов диагностики, таких как МРТ, УЗИ и КТ. Благодаря данным процедурам определяется визуальное увеличение селезёнки и печени (сплено- и ), а также повреждение структуры подкорковых нейронных узлов коры головного мозга.
В обязательном порядке проводится генетическая часть диагностики заболевания, которая заключается в тестировании на генном уровне крови самого пациента и его ближайших родственников. Данное исследование позволяет определить наличие в организме патологического гена и возможную предрасположенность.
Лечение
Лечение болезни Вильсона-Коновалова носит симптоматичный характер и в его основу входит снижение запасов меди в организме и уменьшение её поступления с продуктами питания.
Медикаментозное лечение направлено на вывод меди из организма при помощи соответствующих лекарственных препаратов: соли цинка и D-пеницилламина. Каждый пациент лечится по индивидуальной схеме, что очень важно при данном заболевании. Постепенно дозы препаратов увеличиваются, что позволяет добиться лучшего результата.
Составной частью терапии является регулярное посещение специалиста, не реже одного раза в шесть месяцев, на протяжении которых проводится ряд необходимых обследований, в том числе и мониторинга почек и печени. В случае развития печёночной недостаточности производится хирургическое лечение, которое подразумевает трансплантацию здоровой печени. При удачной приживаемости органа пациент считается полностью здоровым и не нуждается в проведении пожизненной терапии.
Осложнения
Осложнения болезни Вильсона-Коновалова при ненадлежащем или несвоевременном лечении имеют масштабный характер. Наблюдаются нарушения во многих системах и органах:
- Кожа – повышается пигментации, появляются голубые лунки на ногтевых пластинах;
- Почки – появляются почечные камни, что сопровождается отёчностью голеней;
- Сердце – нарушается ритм;
- Суставы – в 20-50% случаев наблюдается поражение суставной ткани;
- Кости – повышается хрупкость, учащаются переломы;
- Эндокринная система - происходит задержка полового развития, у мужчин увеличиваются грудные железы, а у женщин возникают проблемы с вынашиванием плода.
Прогноз
Зависит от сроков и длительности лечения. Благоприятным прогноз считается при своевременном диагностировании и лечении болезни.
Передача дефектного гена по наследству носит аутомно-рецессивный характер. То есть если у обоих родителей данный ген отсутствует, то и у ребёнка его не будет. Если же у двоих родителей присутствует дефектный ген, то при каждой беременности вероятность рождения детей с данным заболеванием составляет 25%. При этом в половине случаев рождаются носители патологического гена, без видимых симптомов заболевания и в 1/4 случаев – полностью здоровые дети, в организме которых патологический ген отсутствует.
Именно поэтому если у родителей уже имеется ребёнок с заболеванием Вильсона-Коновалова, то планировать следующего ребёнка следует под строгим контролем специалистов в данной области, что даёт шанс появления на свет здорового потомства.
Питание и диета
Человек, у которого имеется в организме дефектный ген, должен на протяжении всей жизни придерживаться строгой диеты, предполагающей полное исключение продуктов, богатых данным элементом:
- Мясо - свинина, баранина, мясо уток, фазана, гусей;
- Субпродукты – почки, сердце, печень;
- Рыба – сёмга;
- Морепродукты – кальмары, креветки, устрицы, лобстер, мидии, крабы, морские гребешки;
- Сухофрукты – финики, изюм, чернослив;
- Фрукты – авокадо;
- Бобовые – фасоль, чечевица, горох;
- Овощи – картофель;
- Соевые продукты, молочный шоколад, ячмень, ржаной хлеб, минеральная вода, какао.
Как мы экономим на добавках и витаминах : витамины, пробиотики, муку без глютена и пр. и мы заказываем на iHerb (по ссылке скидка 5$). Доставка в Москву всего 1-2 недели. Многое дешевле в несколько раз, нежели брать в российском магазине, а некоторые товары в принципе не найти в России.
В настоящее время среди молодежи нередко наблюдаются различные расстройства в поведении. У одних проявляется гиперактивность , у других – расстройство внимания , у третьих – гиперсексуальность либо эмоциональная неустойчивость , у четвертых – некритичность . Такие нарушения в поведении родители молодых людей, а вслед за ними – и врачи довольно часто объясняют «переходным возрастом », который по-научному принято называть пубертатным периодом . Такой период, как правило, проявляется у девочек в возрасте от 12 до 16 лет, и у молодых людей в возрасте от 13 до 17 лет.
Если время проходит, пубертатный период у девушек и юношей заканчивается, а признаки «болезни переходного возраста» остаются, либо напротив, усиливаются, это является одним из симптомов уже другого, более серьезного заболевания. Как правило, болезнь вызывает снижение успеваемости как в школе, так потом и в ВУЗе, особенно на первых курсах, а родителям с каждым днем все сложнее наладить контакт со своим уже подросшим «чадом». При этом молодые люди и девушки нередко сквернословят и грубят старшим, довольно часто общаются на повышенных тонах, порой срываются в крик, а ситуация может осложниться уходом подростка из дома.
Если при этом речь молодого человека зачастую становится невнятной, наблюдается нарушенная координация движений, нехарактерное дрожание тела, проявляющаяся в форме причудливых движений конечностями, такие симптомы уже показывают необходимость скорейшего обращения к невропатологу .
Данное заболевание впервые было диагностировано в начале ХХ столетия, в западном мире его симптоматику первым описал ученый и исследователь А.К. Вильсон , а в отечественной медицине исследованиями столь необычного неврологического заболевания занимался Н. А. Коновалов , по фамилии которого и была названа такая болезнь. Патологический ген, появляющийся в организме больного и провоцирующий болезнь Вильсона-Коновалова , был обнаружен сравнительно недавно, лишь в 1993 году.
Заболевание, изученное Вильсоном и Коноваловым, связано с , которая зачастую приводит к понижению в крови концентрации белка церрулоплазмина , отвечающего за переноску меди . Это приводит к недостаточному снабжению медью каждой клеточки организма, и ее неравномерной концентрации в той или иной части тела. Если в организме человека накапливается чрезмерное количество меди, от этого страдают чечевицеобразные ядра промежуточного мозга и .
Симптомы синдрома Вильсона-Коновалова
Проявление болезни Вильсона-Коновалова может быть двояким, особенно существенно различается дебют в каждой из двух форм. Если патология затрагивает печень, у больного впоследствии может развиться , а при неврологической форме синдрома Вильсона-Коновалова поражается мозжечок и кора головного мозга , базальные ганглии . Для неврологической формы заболевания Вильсона-Коновалова характерны проявления и гримасничанья, хореиформные движения конечностями, возникновение атаксии, у некоторых пациентов нередко наблюдаются эпилептические припадки .
Вариант болезни Вильсона-Коновалова, при котором поражается печень, как правило, начинает развиваться у подростков только с одиннадцатилетнего возраста. Неврологическая форма обычно проявляется после девятнадцати лет, когда пубертатный период уже заканчивается, но у некоторых пациентов болезнь Вильсона-Коновалова развивается уже не в молодом, а в пенсионном либо .
Неврологические и печеночные нарушения при этом проявляются относительно с одинаковой частотой, а при отсутствии современного медицинского лечения у больных стремительно развиваются обе формы патологии. Как правило, для начальной стадии болезни Вильсона-Коновалова, характерны лишь психические нарушения , но при этом даже долгое лечение у психиатров оказывается неэффективным и безрезультатным. Довольно часто дебют заболевания характеризуют такие симптомы синдрома Вильсона-Коновалова как и , возникновение гемолитической анемии .
Переизбыток меди в организме, характерный для синдрома Вильсона-Коновалова, вызывает такие сопутствующие заболевания, как и , – патологических изменений сосудов, синдром Фанкони , проявления которого напоминают , при этом атеросклероз развивается ускоренными темпами.
Диагностика синдрома Вильсона-Коновалова
Для выявления синдрома Вильсона-Коновалова нужно сделать развернутый анализ крови , чтобы проверить уровень содержания в организме меди , церулоплазмина , цинка . Но для качественной диагностики анализа крови недостаточно, так как нередко вся накопившаяся в организме медь скапливается не в крови, а глубоко в тканях, а в составе крови ее количество, напротив, существенно ниже нормы. У больных, страдающих печеночной формой синдрома Вильсона-Коновалова, нередко выделяются так называемые печеночные , если к тому моменту уже наблюдается нарушение в работе почек. Симптомы синдрома Вильсона-Коновалова проявляются и зрительными нарушениями появление роговичных колец , наличие которых может выявить лишь офтальмолог, свидетельствует о переизбытке меди в организме.
Лечение синдрома Вильсона-Коновалова
Для лечения синдрома Вильсона-Коновалова сейчас активно используется D —пеницилламин , известный и под названием . Эффективность лечения возможна лишь при терапии, направленной на снижение меди в организме, начатой на ранней стадии развития синдрома Вильсона-Коновалова , при запущенных формах данное заболевание полноценному лечению не поддается.
Для профилактики ее дальнейшего развития необходим регулярный мониторинг, позволяющий правильно определить содержание в организме цинка, меди и уровня церулоплазмина. Необходима регулярная диагностика синдрома Вильсона-Коновалова, обследование у врача должно сопровождать своевременным забором крови на
СЗГМУ им. И.И. Мечникова
Кафедра неврологии
Тема: Болезнь Вильсона-Коновалова.
Составил студент 4 курса
Медико-профилактического факультета
402 группы - Круглов Степан Сергеевич
Преподаватель: Зуев Андрей Александрович
Санкт-Петербург 2013г.
Болезнь Вильсона-Коновалова
Болезнь Вильсона-Коновалова (или гепатоцеребральная дистрофия) - редкое наследственное заболевание, в основе которого лежит генетически обусловленное нарушение обмена меди с избыточным ее накоплением преимущественно в печени и нервной системе. Описано в 1883 г. Вестфалем и в 1912 г. Вильсоном. Термин «гепатоцеребральная дистрофия» предложен Н.В.Коноваловым.
Этиология и патогенез.
В основе лежит аутосомно-рецессивное наследственное нарушение метаболизма меди; ген расположен в длинной части хромосомы 13. Распространенность в различных регионах мира в среднем 1:30000 при частоте гетерозиготного носительства около 1 %.
Первоначально ген экспрессируется в печени, почках, плаценте. Продукт гена представляет собой катионтранспортирующий Р-тип АТФазного протеина. Следствием генетического дефекта является различной степени выраженности нарушение функции внутриклеточного транспорта меди. Это ведет к снижению экскреции меди с желчью и накоплению её в гепатоцитах.
С пищей в сутки поступает 2-5 мг меди. Она всасывается в кишечнике, поступает в печень, где связывается с синтезируемым печенью церулоплазмином, циркулирует в сыворотке крови, избирательно захватывается органами и экскретируется с желчью.
В норме экскреция меди с желчью 2 мг в сутки, при болезни Вильсона-Коновалова - только 0.2-0.4 мг, что приводит к повышенному накоплению меди в организме.
Включение меди в церулоплазмин происходит в аппарате Гольджи при участии гена гепатоцеребральной дистрофии. Незначительная часть меди находится в крови в ионизированной форме в виде лабильного комплекса с альбумином и выделяется с мочой.
При болезни Вильсона-Коновалова увеличена абсорбция меди в кишечнике, снижена экскреция меди с желчью. Снижение экскреции меди связано с дефектом гена гепатоцеребральной дистрофии, определяющего транспорт меди в аппарат Гольджи и последующее выделение лизосомами в желчь. Нарушается процесс включения меди в церулоплазмин. Из-за недостаточного использования меди происходит её депонирование в печени, мозге, почках, роговице. Депонированная в печени медь вторично ингибирует синтез церулоплазмина.
Уровень церулоплазмина в сыворотке крови имеет диагностическое, но не патогенетическое значение. У 5 % больных определяется нормальный уровень церулоплазмина. При биопсии печени у таких больных имеется избыточное количество меди, также увеличивается содержание меди в крови и тканях, выделение её с мочой.
Медь, являясь прооксидантом, оказывает токсическое действие на организм. Её накопление ведет к повышенной продукции свободных гидроксильных радикалов. При обследовании больных болезнью Вильсона-Коновалова и животных с экспериментальной перегрузкой медью в плазме крови определяется снижение уровня витамина Е, увеличение циркулирующих продуктов перекисного окисления липидов; в печени снижены уровни восстановленного глутатиона и витамина Е.
Митохондрии печени являются мишенями действия оксидантов. Нарушение дыхательной цепи и снижение активности цитохром-С-оксидазы увеличивает продукцию свободных радикалов благодаря утечке электронов из дыхательной цепи.
Свободная медь, накапливаясь в тканях, блокирует SH-группы ферментов, участвующих в окислительно-восстановительных реакциях. Это приводит к энергетическому голоданию, к которому наиболее чувствительна ЦНС.
В начале заболевания, когда клинические признаки отсутствуют (Iстадия), медь накапливается в цитозоле печеночных клеток. Медь, связанная сSH-группами цитозольных протеинов, затрудняет секрецию гепатоцитами белков и триглицеридов. Наступает стеатоз гепатоцитов и появление телец Маллори.
Во IIстадии медь перераспределяется из цитозоля в лизосомы гепатоцитов. Часть поступает в кровь. В связи с низкой специфической активностью лизосом билиарная экскреция меди понижается. Медь вызывает переокисление липидов и повреждение лизосомальных мембран с последующим выходом вредных кислых гидролаз в цитоплазму. Наблюдаются некроз гепатоцитов, развитие хронического гепатита и гемолитической анемии.
В IIIстадии усиленное накопление меди в печени приводит к фиброзу и циррозу. Повышенное накопление меди в головном мозге, роговице, дистальных отделах почечных канальцев приводит к развернутой картине болезни.
Морфология.
В печеночной ткани наблюдаются жировая дистрофия гепатоцитов, перипортальный фиброз, субмассивные некрозы гепатоцитов, макронодуллярный цирроз. В почках - жировая и гидропическая дистрофия с отложением меди в проксимальных канальцах.
Клиническая картина.
Клинические проявления:
Печеночные – цирроз печени, хронический активный гепатит, фульминантная печеночная недостаточность.
На начальной стадии изменения в печени неспецифические - мелко- и среднекапельная жировая дистрофия, некрозы единичных гепатоцитов, перипортальный фиброз. Далее развивается клиника хронического гепатита высокой степени активности с желтухой, высоким уровнем аминотрансфераз, гипергаммаглобулинемией. При прогрессировании - цирроз печени с портальной гипертензией и печеночноклеточной недостаточностью.
Фульминантная печеночная недостаточность - редкое проявление гепатоцеребральной дистрофии. Развивается у подростков и молодых людей. Характерные признаки, позволяющие дифференцировать её с фульминантным гепатитом вирусной этиологии: небольшое повышение активности трансаминаз (с преобладанием повышения активности АСТ), низкий уровень щелочной фосфатазы, крайне низкий уровень альбумина в сыворотке крови, высокий уровень прямого и непрямого билирубина (внутрисосудистый гемолиз), гемоглобинурия, высокий уровень меди в сыворотке печени и её экскреции с мочой. Часто сопровождается гемолитической анемией, связанной с массивным высвобождением меди из печени.
Существует абдоминальная форма Керара - поражение печени преобладает на всем протяжении болезни и рано осложняется печеночной недостаточностью. В дебюте – развитие отечно-асцитического синдрома, степень выраженности которого не соответствует выраженности других признаков портальной гипертензии. Постоянное наличие большого количества несвязанной меди в сыворотке крови и повышенное отложение её не только в печени, но и в других органах приводит к повреждению головного мозга, роговицы, почек, скелета, гемолизу эритроцитов.
Неврологические – экстрапирамидные, церебеллярные, псевдобульбарные нарушения, судорожные припадки.
Две основные формы заболевания - ригидно-аритмогиперкинетическая, или ранняя, и дрожательная - значительно различаются по своим клиническим проявлениям. Первая характеризуется быстрым развитием общей ригидности и наличием неритмичных гиперкинезов атетоидного или торсионно-спастического характера. Ригидность распространяется на мышцы туловища, конечностей и на мышцы, участвующие в глотании и речевом акте. Отмечаются амимия, дисфагия, дизартрия. Походка становится скованной, подпрыгивающей. Ригидность мышц может приступообразно усиливаться, особенно в связи с произвольными движениями и под влиянием эмоций. Больные часто застывают в самых неудобных позах. В дистальных отделах конечностей нередко образуются контрактуры. Нарастающая ригидность быстро приводит к полной обездвиженности. Эта форма заболевания начинается в детском возрасте - от 7 до 15 лет. Висцеральные расстройства могут проявляться раньше - в возрасте 3-5 лет. Как правило, выражены признаки печеночной патологии, которые часто предшествуют развитию неврологической симптоматики.
Флексорно-экстензорный тремор. Его выраженность колеблется от едва заметного дрожания рук до тремора всего тела. Тремор усиливается при волнении и целенаправленных действиях. Умеренный тремор у ряда больных может иметь акцент на одной стороне. Тремор пальцев вытянутых рук типичный, «порхающий».
Мышечная дистония наблюдается у всех больных. Проявления дрожательно-ригидной формы различной выраженности. Определяется гипомимия, гиперсаливация, затрудненная монотонная речь, снижение интеллекта. Акинетико-ригидная форма сопровождается ярко выраженным ригидным синдромом, затрагивающим различные группы мышц. В развернутой стадии выявляется гиперкинез по типу «бьющихся крыльев», к которому могут присоединяться интенционный компонент, дизартрия, дисфагия, мозжечковые расстройства, миоклонии. Без специфической терапии нарастание симптоматики приводит к выраженным контрактурам, обездвиженности, грубой деменции.
У больных с экстрапирамидной патологией могут развиваться пирамидные моно- и гемипарезы. Такие случаи относятся к экстрапирамидно-корковой форме гепатоцеребральной дистрофии, которая отличается от других форм значительным поражением коры больших полушарий. У больных часто отмечаются эпилептические припадки общего и особенно джексоновского характера, тяжелое нарушение интеллекта с грубыми нарушениям личности. Психические нарушения могут иметь место и у больных с другими формами заболевания. Они характеризуются изменениями эмоционально-волевой сферы, снижением психической активности и интеллекта. Наряду с этим наблюдаются случаи доброкачественного течения гепатоцеребральной дистрофии, когда у больных в течение длительного времени неврологическая симптоматика отсутствует или имеются очень легкие симптомы, которые не нарушают их трудоспособности. Такие больные, как правило, выявляются случайно при обследовании семей больных, с развернутой картиной заболевания.
Психиатрические - нарушения в эмоциональной сфере, психоз, нарушения поведения, познавательной деятельности.
Гематологические - гемолиз, анемия, тромбоцитопения, нарушения свертывающей системы крови. У 15 % больных могут наблюдаться явления острого внутрисосудистого гемолиза. Гемолиз обычно временный, проходит самостоятельно, предшествуя ярким клиническим признакам поражения печени в течение нескольких лет. Иногда может протекать одновременно с острой печеночной недостаточностью. Предполагается влияние больших количеств свободной меди в плазме на мембраны эритроцитов и гемоглобин.
Почечные - канальцевые нарушения (частичный или полный синдром Фанкони), снижение клубочковой фильтрации, нефролитиаз.
Поражение почек проявляется периферическими отеками, микрогематурией, незначительной протеинурией, повышением концентрации креатинина сыворотки крови. Как ранний симптом может наблюдаться макро- и микрогематурия. Наиболее часто в моче обнаруживают треонин, тирозин, лизин, валин, фенилаланин.
Офтальмологические - кольцо Кайзера-Флейшера, катаракта (содержащие медь отложения в капсуле хрусталика).
Эндокринологические - аменорея, спонтанные аборты, задержка полового развития, гинекомастия, гирсутизм, ожирение, гипопаратироидизм.
Сердечно-сосудистые - кардиомиопатия, аритмия.
Мышечно-скелетные - остеомаляция, остеопороз, артропатия, артралгии.
Желудочно-кишечные - холелитиаз, панкреатит, спонтанный бактериальный перитонит.
Дерматологические - голубые лунки у ногтевого ложа, сосудистая пурпура, гиперпигментация кожи,acantosisnigricans.
Сам Н.В.Коновалов выдел пять форм: брюшная форма, ригидноаритмогиперкинетическая, или ранняя форма, дрожательно-ригидная форма, дрожательная форма, экстрапирамидно-корковая форма. ГЛД может начать проявляться в детском, подростковом, юношеском, зрелом возрасте и очень редко - в 50-60 лет. В 40-50 % случаев заболевание манифестирует с поражения печени, в 35-50% - с различных неврологических и/или психиатрических расстройств.
Брюшная форма - характеризуется манифестацией заболевания до 40 лет и тяжелым поражением печени по типу хронического гепатита; цирроза печени; быстропрогрессирующего (фульминатного) гепатита.
Ригидно-аритмогиперкинетическая - дебютирует в детском возрасте. Начальными симптомами могут быть трудности при выполнении мелких движений, отмечается мышечная ригидность, брадикинезия, амимия, смазанность речи, нередки эпилептические приступы, психиатрические расстройства и умеренное снижение интеллекта; Течение заболевания прогрессирующее, с эпизодами обострений и ремиссий. Дрожательно-ригидная форма - одна из наиболее частых форм ГЛД, с пиком манифестации в ювенильном возрасте. Основными и ведущими симптомами являются мышечная ригидность и тремор, усиливающийся при физическом напряжении и исчезающий во сне. В некоторых случаях наблюдаются атетоидные и хореиформные гиперкинезы, расстройства глотания и речи.
Дрожательная форма - начинается на втором-третьем десятилетии жизни. В клинической картине преобладает тремор. Частыми симптомами являются брадилалия, брадикинезия, тяжелый психо-органический синдром, нередки эпилептические приступы. Экстрапирамидно-корковая форма - встречается реже других форм, начинается обычно как одна из вышеописанных форм. Типичные для данной формы ГЛД экстрапирамидные и пирамидные нарушения, эпилептические приступы и выраженный интеллектуальный дефицит.
Диагностика.
Подозрение на наличие болезни Вильсона-Коновалова должно возникнуть при:
неуточненной этиологии хронического гепатита и цирроза;
фульминантной печеночной недостаточности;
необъяснимом повышении уровня аминотрансфераз;
наличии соответствующих неврологических изменений неустановленной этиологии, изменении поведения;
психических симптомах, сочетающихся с признаками заболевания печени;
необъяснимой приобретенной гемолитической анемии;
семейном анамнезе по гепатоцеребральной дистрофии.
Основные (скрининговые) тесты для диагностики болезни Вильсона-Коновалова:
обнаружение кольца Кайзера-Флейшнера: не обнаруживается у 50-62 % больных без неврологических симптомов; может отсутствовать у 5 % больных с начальными признаками поражения ЦНС;
выявление снижения содержания церулоплазмина в сыворотке крови до уровня <20 мг/дл (норма 25-50 мг/дл): уровень <5 мг/дл - абсолютное доказательство болезни Вильсона-Коновалова. Умеренное снижение может встречаться у гетерозиготных носителей гена, при циррозе печени другой этиологии, при синдроме мальабсорбции, нефротическом синдроме и др.; у 10-15 % больных с абдоминальной формой заболевания уровень церулоплазмина может быть в пределах нормы;
увеличение содержания не связанной с церулоплазмином меди в сыворотке крови (300 мкг/л и >);
повышение экскреции меди с мочой (более 200 мкг в сутки при норме < 70 мкг в сутки);
D-пеницилламиновый тест - повышение суточной экскреции меди до уровня >1500 мкг, в норме значительного увеличения экскреции меди с мочой не наблюдается;
высокий уровень включения изотопа меди в церулоплазмин - в норме - отсутствие пика включения через 48 часов; тест диагностически значим только у больных с нормальным уровнем церулоплазмина;
генетические исследования: значимы у сибсов и других членов семьи пробанда.
Для количественного определения меди в биоптатах печени используют спектрофотометрию, рентгеноструктурный анализ.
Также для диагностики используют поглощение печенью радиоактивной меди. Соотношение радиоактивности печени через 24 и 2 часа после внутривенного введения радионуклида меди в норме равно 1.4-9, а при болезни Вильсона-Коновалова 0.2-0.3. Гетерозиготные носители и больные с другими заболеваниями печени имеют соотношение, равное единице. Кинетика радиоактивной меди позволяет дифференцировать болезнь Вильсона-Коновалова от гепатоцеребрального синдрома при заболеваниях печени.
При КТ/МРТ головного мозга визуализируют атрофию полушарий большого мозга и мозжечка, базальных ядер, в некоторых случаях - очаги некрозов в проекции скорлупы.
Лечение.
Терапия направлена на выведение избытка меди из организма для предупреждения её токсического воздействия.
Назначают диету № 5, богатую белком, с ограничением содержащих медь продуктов (баранина, куры, утки, колбасы, рыба, ракообразные, шампиньоны, кресс-салат, щавель, лук-порей, редис, бобовые, орехи, чернослив, каштаны, шоколад, какао, мед, перец и др.).
Основа терапии - использование препаратов, связывающих медь и выводящих её из организма:
Британский антилюизит (2,3-димеркаптопропанол) - вводят внутримышечно по 1.25-2.5 мг/кг 2 раза в день в течение 10-20 дней, перерыв между курсами 20 дней. Другая методика применения: введение 200-300 мг 2 раза в день в течение нескольких месяцев до получения эффекта. Применение препарата ограничено из-за болезненности инъекций и появления признаков интоксикации при длительном лечении.
Унитиол 5% - по 5-10 мл ежедневно или через день, на курс 25-30 внутримышечных инъекций. Повторные курсы через 2-3 месяца.
D -пеницилламин. Увеличивает выведение меди с мочой: образует комплексы, которые легко фильтруются через почечные клубочки. Дозы от 0.3-1.3 до 3-4 граммов в сутки в зависимости от величины экскреции меди с мочой. Оптимальная доза препарата 0.9-1.2 грамма в сутки.
Доза препарата должна устанавливаться ежегодно, а при длительном лечении каждые 2 года на основании выделения меди с мочой, контрольных биопсий печени и определения содержания меди в биоптатах печени.
Клиническое улучшение под влиянием лечения выражается в сглаживании неврологической симптоматики, снижении активности воспалительного процесса в печени. При успешной терапии D-пеницилламином выведение меди с мочой увеличивается в 3-5 раз. В первые 2-3 недели от начала лечения может наблюдаться усиление неврологической симптоматики и ухудшение функционального состояния печени, которое затем сменяется улучшением, обычно через несколько недель или месяцев. Есть описания полного исчезновения активности хронического гепатита и цирроза по данным биопсии печени спустя годы после применения препарата.
Трансплантация печени.
Показана при фульминантной печеночной недостаточности, прогрессировании печеночной недостаточности на фоне хронического гепатита и цирроза печени при неэффективности медикаментозной терапии.
Прогноз.
Течение заболевания прогрессирующее, ведущее к инвалидизации. Прогноз улучшается при назначении адекватной терапии на ранних стадиях заболевания. Терапия на поздней стадии существенно не влияет на развитие осложнений.
Смерть наступает преимущественно в молодом возрасте, как правило, от осложнений цирроза печени (кровотечения из варикознорасширенных вен пищевода, печеночная недостаточность) или фульминантного гепатита, реже - от осложнений, связанных с поражением ЦНС.
Профилактика.
Ранняя диагностика заболевания. При выявлении дефектного гена в гомозиготном состоянии лечение медьхелатирующими препаратами может быть начато в раннем детском возрасте.
Литература:
С.Д. Подымова. Болезни печени: Руководство. – 4 издание, переработанное и дополненное. – М.: ОАО «Издательство «Медицина», 2005. – 768 с. (сс. 567-578).
Ш. Шерлок, Дж. Дули. Заболевания печени и желчных путей: Практическое руководство.: Перевод с английского./Под редакцией З.Г. Апросиной, Н.А. Мухина. – М.: Гэотар Медицина, 1999. – 864 с. (сс. 476-483).
Справочник практического врача по гастроэнтерологии./Под редакцией В.Т. Ивашкина, С.И. Рапопорта. – М.: Советский спорт, 1999. – 432 с. (сс. 175-177).
Справочник Харрисона по внутренним болезням./Под редакцией К. Иссельбахера, Е. Браунвальда, Дж. Вильсон и др. – СПб: Издательство «Питер», 1999. – 976 с. (сс. 786-787).
Т.М. Игнатова. Ранняя диагностика болезни Вильсона-Коновалова: радикальное улучшение прогноза. Врач, 2004, № 12, сс. 36-39.
Сыр Сулугуни относится к твердым сортам с использованием рассола. Этот продукт имеет достаточно плотную консистенцию, которая может быть белого или же кремового цвета (см. фото). Родиной для сыра Сулугуни является Грузия. Для приготовления используют овечье, буйволиное, козье и коровье молоко, которое прошло пастеризацию.
Технология производства этого сыра достаточно необычная. Сначала молоко смешивают с бактериями, хлористым кальцием, пепсином, и потом нагревают примерно до 36 градусов. Полученный сыр прессуют и оставляют на несколько часов в серпянке при увеличенной температуре. Следующий шаг – массу делят на кубики и плавят в кислой воде или же сыворотке. Затем сырную массу мешают, чтобы в итоге она стала тягучей и однородной. После этого все кладут на стол, делят на кусочки и собирают все в шары. Затем головки кладут в формы, которые имеют цилиндрическую форму, а далее их выдерживают пару суток в рассоле с низкой температурой.
Готовый продукт обладает плотной консистенцией, которая имеет слои, небольшие пустоты и глазки. Кроме этого, у этого сыра нет корки, и на его поверхности иногда можно увидеть легкую слоистость. Качественный продукт должен быть равномерно окрашен в белый цвет, также иногда может присутствовать легкий желтый оттенок.
Сыр Сулугуни можно приобрести в совершенно разных формах, к примеру, в брусочках, но наибольшей популярностью пользуются сырные косички. Кроме этого, сегодня сыр подкапчивают, что позволяет увеличить срок хранения и придать необычный вкус.
Выбор и хранение
 При выборе сыра Сулугуни стоит обращать внимание на цвет и консистенцию продукта.
Если его неправильно или уже достаточно долго хранят, у продукта будет отсутствовать характерный запах и вкус. О том, что сыр Сулугуни испортился, свидетельствует наличие сухой корки, трещин и плесени.
При выборе сыра Сулугуни стоит обращать внимание на цвет и консистенцию продукта.
Если его неправильно или уже достаточно долго хранят, у продукта будет отсутствовать характерный запах и вкус. О том, что сыр Сулугуни испортился, свидетельствует наличие сухой корки, трещин и плесени.
Хранить сыр Сулугуни нужно в рассоле. В холодильнике продукт будет сохранять свою свежесть в течение 3-х месяцев. Чтобы продлить срок хранения, сначала на некоторое время продукт нужно положить в свежее молоко, а после в концентрированный рассол (400 г соли на 1 л воды). В таком виде сыр должен находиться в течение дня. После его нужно положить в другой рассол (200 г соли на 1 л воды).
Польза сыра Сулугуни
 Польза сыра Сулугуни обусловлена наличием большого количества витаминов и минералов.
Этот продукт позволяет быстро восстановить энергетические затраты организма, а также улучшить обмен веществ. Кроме этого, при регулярном потреблении продукт способствует улучшению метаболизма в клетках.
Польза сыра Сулугуни обусловлена наличием большого количества витаминов и минералов.
Этот продукт позволяет быстро восстановить энергетические затраты организма, а также улучшить обмен веществ. Кроме этого, при регулярном потреблении продукт способствует улучшению метаболизма в клетках.
Есть в сыре Сулугуни сера, которая снижает уровень холестерина в крови, а также она принимает активное участие в обменных процессах в организме. В большом количестве содержится в этом продукте натрий, который необходим для нормализации водного баланса в организме. Есть в этом сыре фосфор, который принимает активное участие в синтезе белка и в формировании костной и мышечной ткани. Благодаря калию нормализуется деятельность сердечно-сосудистой системы. Входит в состав Сулугуни и кальций, который укрепляет кости, зубы и ногти.
Использование в кулинарии
Сыр Сулугуни прекрасно сочетается с многочисленными продуктами. К примеру, его достаточно часто подают к вину с травами. Также этот сыр входит в рецепты разнообразных овощных салатов. Этот сыр – прекрасная начинка для разнообразной выпечки. Кроме этого, Сулугуни кладут в суфле, паштет, омлет, пиццу и в другие блюда.
Вред сыра сулугуни и калорийность
Вред сыр Сулугуни может принести людям с индивидуальной непереносимостью продукта. Копченые сорта не стоит употреблять при гастрите, язве, а также при почечных и сердечных отеках. Стоит также учитывать высокую калорийность, а значит, его не рекомендуется употреблять в период похудения и при ожирении.
Грузинский твердый рассольный сыр, который начали производить в регионе Самегрело. Отличается молочным вкусом, немного солоноватым, приятным, нежным запахом и податливой, упругой, слоистой консистенцией. По объему сырного тела могут располагаться небольшие глазки. Сулугуни не имеет корочки, а его цвет в зависимости от молока, из которого он произведен, может быть от белоснежного до светло-желтого. Выпускается также копченый Сулугуни.
Сыр может производиться в виде цилиндрических головок или косичек.
Относительно происхождения названия сыра существует несколько версий. По одной из них, осетинское слово (дигорский диалект) «сулугун» означает «созданный из сыворотки». Осетины, живущие в Дигорском ущелье, называют его «сулугун». Согласно другой версии имя сыра образовано двумя словами: «сули» (душа) и «гули» (сердце), что может означать «с душой и сердцем». Наличие Сулугуни на столе издавна свидетельствовало о гостеприимности и достатке хозяев.
В сентябре 2011 года правительство Грузии оформило патент на Сулугуни.
Изготовление
Для производства 1 кг сыра берут около 10 л молока. При этом может использоваться коровье или буйволиное молоко, а также их смесь. Молоко пастеризуют, а затем вносят молочнокислую закваску, хлористый кальций и стрептококки, отвечающие за аромат. При температуре 31-35 градусов добавляют сычужный фермент и пепсин. Процесс коагуляции продолжается 30-35 минут. После этого калье разрезают на кубики со стороной от 6 до 10 мм и дают массе 5-10 минут отдохнуть. Затем сыр обсушивается на протяжении 10-20 минут.
Сулугуни повторно нагревают в течение 10-15 минут до 34-37 градусов. После этого из сырной массы удаляют сыворотку, кубики формируют в пласт и немного прессуют. Затем будущий Сулугуни подвергается чеддеризации в большой ванне при температуре сыворотки 28-32 градуса. Эта стадия продолжается от 2 до 3 часов. Степень готовности сыра определяют плавлением: небольшой кусочек размерами примерно 0,8х12 см на 1-2 минуты помещают в воду температурой 90-95 градусов и вытягивают, формируя тонкие нити. Если они не разрываются, то чеддеризация завершена.
С помощью шпигорезки сырную массу разрезают на кусочки размерами около 1,3-2,5 см и помещают в воду или сыворотку, нагретую до температуры 70-80 градусов. Массу хорошо вымешивают до получения тягучей консистенции. Затем сыр перекладывают на стол и приступают к формовке. Для этого отрезают кусок нужного размера и несколько раз заворачивают его края в середину, в итоге получая цилиндрическую головку, которую помещают в форму.
Сформировавшиеся головки солят в кисло-сывороточном рассоле при температуре 8-12 градусов на протяжении 6-48 часов.
С чем сочетается
Сулугуни может быть сырым, копченым, печеным, жаренным. Его подают к вину вместе с кинзой и базиликом, добавляют в салаты, хачапури, пироги, выпечку, вторые блюда.
Сыр сочетается с фасолью, маслинами, огурцами, красным луком, редисом, мясными и рыбными блюдами, оливковым маслом, бальзамическим уксусом, сметаной, разнообразными соусами, горчицей.



